Abstract
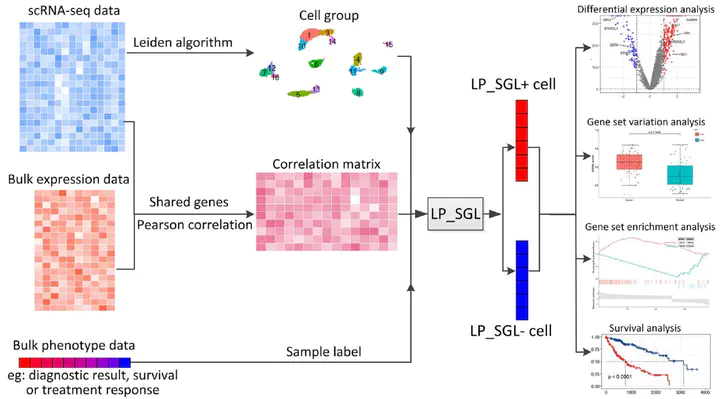
Single-cell RNA sequencing (scRNA-seq) enables the resolution of cellular heterogeneity in diseases and facilitates the identification of novel cell types and subtypes. However, the grouping effects caused by cell–cell interactions are often overlooked in the development of tools for identifying subpopulations. We proposed LP_SGL which incorporates cell group structure to identify phenotype-associated subpopulations by integrating scRNA-seq, bulk expression and bulk phenotype data. Cell groups from scRNA-seq data were obtained by the Leiden algorithm, which facilitates the identification of subpopulations and improves model robustness. LP_SGL identified a higher percentage of cancer cells, T cells and tumor-associated cells than Scissor and scAB on lung adenocarcinoma diagnosis, melanoma drug response and liver cancer survival datasets, respectively. Biological analysis on three original datasets and four independent external validation sets demonstrated that the signaling genes of this cell subset can predict cancer, immunotherapy and survival.